
In preclinical experiments, University of Alabama at Birmingham researchers have revealed a key missing piece of the Alzheimer’s disease puzzle. That allowed proof-of-concept experiments—using an existing drug—that dramatically reduced Alzheimer’s pathology and symptoms in two mouse models, potentially offering an immediate treatment for this devastating disease.
The research was published today in the journal Science Translational Medicine. It included human brain tissue analysis and longitudinal clinical data that supported the in vivo mouse model data.
“Our study provides translational insights into mechanisms underlying amyloid-beta protein toxicity, which may have strong implications for future drug design,” said Qin Wang, M.D., Ph.D. “It identifies an amyloid-beta/G protein-coupled receptor interaction that represents an attractive, disease-specific therapeutic target for Alzheimer’s disease.”
Interestingly, the pathologic mechanism found may also explain the failure of numerous Alzheimer’s clinical trials that targeted reduction of the culprit in Alzheimer’s disease—amyloid protein buildup in the brain.
At the UAB School of Medicine, Wang is professor in the Department of Cell, Developmental and Integrative Biology.
It is widely accepted, Wang says, that buildup of amyloid-beta oligomers in the brain acts as a trigger to induce pathological changes in tau protein, and that altered tau protein is the bullet that targets and kills neurons in Alzheimer’s disease. However, the pathway connecting these two was unknown.
Wang and colleagues have found that amyloid-beta oligomers hijack norepinephrine signaling at brain neurons, which falsely redirects this signal to activate a kinase called GSK3-beta. That activated kinase enzyme, in turn, hyper-phosphorylates tau protein, making it toxic for neurons.
This rewiring of the norepinephrine signaling takes place at a cell membrane receptor on the surface of neurons called the alpha-2A adrenergic receptor. This receptor is part of a large family of G protein-coupled receptors that detect molecules outside of a cell and then activate an internal signal that causes a cellular response. While a certain concentration of amyloid-beta oligomers can activate GSK3-beta, the presence of norepinephrine vastly sensitized that activation by up to two orders of magnitude, Wang and colleagues found.
Thus, the UAB researchers speculate that nanomolar concentrations of amyloid-beta oligomers in human brains induce a pathogenic GSK3-beta/tau cascade at the earliest stages of Alzheimer’s disease. This theory suggests why multiple clinical trials to reduce levels of amyloid-beta oligomers in Alzheimer’s disease patients have failed—they cannot reduce the amyloid levels to such low concentrations.
Study details
The alpha-2A adrenergic receptor normally works this way—it has a binding site for the neurotransmitter norepinephrine, and that binding activates a signaling process that mobilizes the brain and body for action. The UAB researchers found that amyloid-beta oligomers bind to a separate site on the alpha-2A adrenergic receptor, distinct from the site for norepinephrine binding. This initiates the pathological hijacking.
Such binding at a second site is called allosteric binding. In G protein-coupled receptors, allosteric ligands are known to often alter signaling of the receptor as part of normal physiology. After the researchers realized the allosteric binding, they searched to see which kinase might be activated by that binding, which is how they identified GSK3-beta.
Some clinical data supports this mechanism. The researchers found that alpha-2A adrenergic receptor from the postmortem prefrontal cortexes of Alzheimer’s disease patients had a significant increase in alpha-2A adrenergic receptor activity, compared with non-demented, low-pathology controls. Also, epidemiologic analysis of cases from the National Alzheimer’s Coordinating Center showed that taking the drug clonidine—an activator of alpha-2A adrenergic receptor used to lower blood pressure—worsened cognitive function in patients with cognitive deficits. Furthermore, the adverse effects of clonidine were stronger in patients with more severe dementia. Clonidine usage had no effect on subjects with normal cognition.
Wang and colleagues tested an existing drug—idazoxan—in a mouse model of Alzheimer’s. Idazoxan is an alpha-2A adrenergic receptor antagonist that has been under investigation in clinical trials for depression. The hypothesis was that idazoxan blockage of alpha-2A adrenergic receptor in the presence of amyloid-beta pathology would show therapeutic potential. That was borne out in Alzheimer’s-model mice.
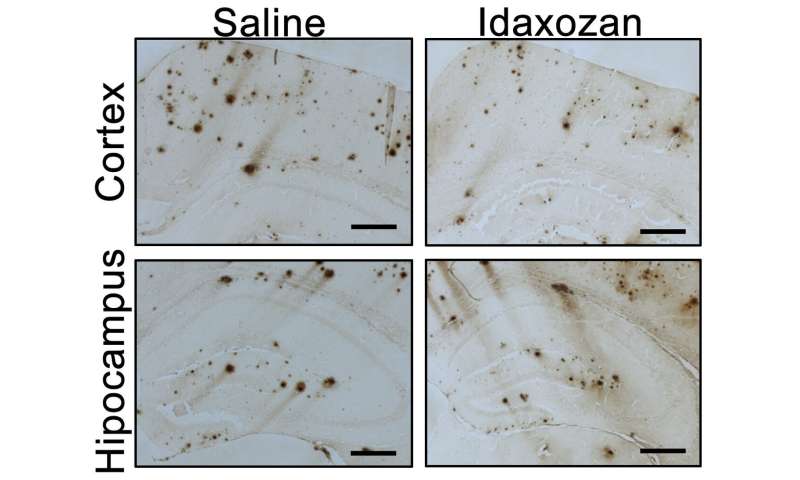
The mice were treated with idazoxan for eight weeks beginning at age 8 months, a point when amyloid-beta plaques are already present in the brain and alpha-2A adrenergic receptor shows enhanced activity. Compared with controls, the UAB researchers found that: 1) Idazoxan reversed hyperactivation of GSK3-beta in the mouse brains, giving additional support for the critical role of alpha-2A adrenergic receptor in mediating amyloid-beta-induced activation of GSK3-beta in vivo; 2) In the cerebral cortex of idazoxan-treated Alzheimer’s-model mice, the extent of amyloid-beta load was lower, showing that blockage of alpha-2A adrenergic receptor slowed the progression of amyloid-beta pathology; 3) Idazoxan treatment decreased the density of inflammatory microglial cells, suggesting a reduction of neuroinflammation; 4) Idazoxan treatment reduced tau hyper-phosphorylation, suggesting that blockade of the alpha-2A adrenergic receptor effectively alleviated amyloid-beta-induced tau pathology; and 5) Idazoxan-treated Alzheimer’s-model mice performed nearly as well as normal mice, and significantly better than untreated Alzheimer’s-model mice, in two tests for cognitive function.
“These data collectively demonstrate that blocking norepinephrine signaling through the alpha-2A adrenergic receptor is an effective strategy to ameliorate pathological and cognitive deficits associated with amyloid-beta,” Wang said.
“The alpha-2A adrenergic receptor blockers such as idazoxan have been developed for use in other disorders, and repurposing these drugs could be a potentially effective, readily available strategy for Alzheimer’s disease treatment,” Wang said. “In addition, our data suggest that the amyloid-beta/alpha-2A adrenergic receptor interaction is an attractive, disease-specific therapeutic target for Alzheimer’s because the alpha-2A adrenergic receptor/GSK3-beta/tau cascade can be activated only in the presence of amyloid-beta oligomers.”
Source: Read Full Article