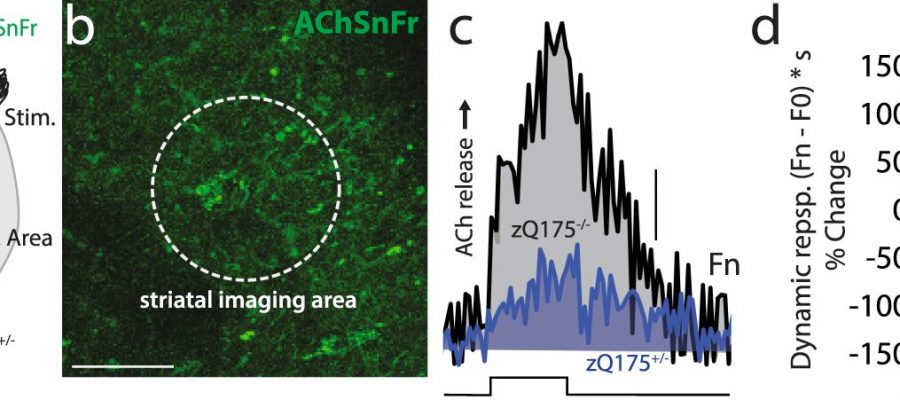
 stereotaxically injected in dorsolateral striatum (DLS). The imaging area and bipolar electrical stimulator are indicated. b DLS section showing the striatal neuropil expressing AAV-AChSnFr and the imaging area (Scale bar: 50 μm). c Representative traces showing a decrease in evoked acetylcholine (ACh) released in zQ175+/− compared to zQ175−/− controls. Scale bar: 0.05 Normalized Fluorescence (Fn). Electrical stimulation (step) consisted of 20 − 1 ms stimuli, of 1 mA delivered at 2 Hz. d Boxplot summarizing the data from zQ175−/− (N = 3; n = 6 slices), zQ175+/− (N = 4; n = 7), **p = 0.0082 Mann–Whitney non-parametric two-sided test. Shown is the % change compared to median data collected in zQ175−/− controls, and the Dynamic response [(Fn – F0) * s], see \"Methods.\" Credit: Nature Communications (2023). DOI: 10.1038/s41467-023-36556-3")
Investigators led by D. James Surmeier, Ph.D., the Nathan Smith Davis Professor and chair of Neuroscience, have uncovered previously unknown neuronal circuits that contribute to brain dysfunction in Huntington’s disease.
The study, published in Nature Communications, prednisone to treat back pain sheds light on novel circuit mechanisms that could serve as potential therapeutic targets for treating patients with Huntington’s.
Huntington’s disease is an inherited, progressive neurodegenerative disease. Common symptoms of Huntington’s include involuntary, hyperkinetic movements and disruptions in behavioral, emotional and cognitive functioning. The movement-related symptoms of the disease are caused by the dysfunction of neurons in the striatum, a subcortical region of the brain involved with habit formation, goal-directed action and voluntary movement.
On the molecular level, patients with Huntington’s disease have an increased number of CAG (Cytosine, Adenine, Guanine) sequence repeats in the huntingtin gene. While the CAG expansion in huntingtin has long been known to cause Huntington’s disease, precisely how the mutant huntingtin protein disrupts neuronal function has remained unresolved.
In the current study, Surmeier’s team used a male mouse model of Huntington’s and optogenetic interrogation of striatal circuits to help fill this gap. They discovered that the synaptic inputs to principal striatal neurons were significantly altered by mutant huntingtin.
Specifically, principal striatal neurons receive information from two different types of cortical neuron: one input from pyramidal tract neurons and the other from intratelencephalic neurons. In the Huntington’s mouse model, the investigators found that the intratelencephalic pathway made stronger connections than in normal mice, whereas those of the pyramidal tract were weaker.
This distortion in information being received by principal striatal neurons was caused by a deficit in the release of acetylcholine by striatal cholinergic interneurons, which are critical to behavioral flexibility or changing behavior in response to certain outcomes.
“When cholinergic interneurons become dysfunctional, striatal circuits have difficulty adjusting to new circumstances. Indeed, this is one of the key characteristics of Huntington’s disease patients: they have difficulty changing their behavior when contingencies change,” Surmeier said.
Next, the investigators used an adeno-associated virus carrying a zinc finger repressor protein to selectively suppress mutant huntingtin. Using this technique, the investigators could suppress the mutated huntingtin gene selectively in striatal cholinergic interneurons, and doing so normalized intratelencephalic connectivity.
“Because mutant huntingtin is widely expressed, the fact that selectively reducing it just in cholinergic interneurons had such a profound effect on striatal connectivity was surprising. This study clearly points to the potential therapeutic value of zinc finger proteins,” Surmeier said.
As for next steps, Surmeier said his team is studying how striatal cholinergic interneurons impact other aspects of striatal circuity and how they might influence involuntary movement in patients with Huntington’s disease.
“The striatal neurons that are particularly vulnerable in Huntington’s disease are involved in inhibition of unwanted actions,” Surmeier said. “We’re now trying to figure out how the cholinergic interneurons are influencing those cells and how they’re involved in normal movement control.”
More information:
Tristano Pancani et al, Cholinergic deficits selectively boost cortical intratelencephalic control of striatum in male Huntington’s disease model mice, Nature Communications (2023). DOI: 10.1038/s41467-023-36556-3
Journal information:
Nature Communications
Source: Read Full Article