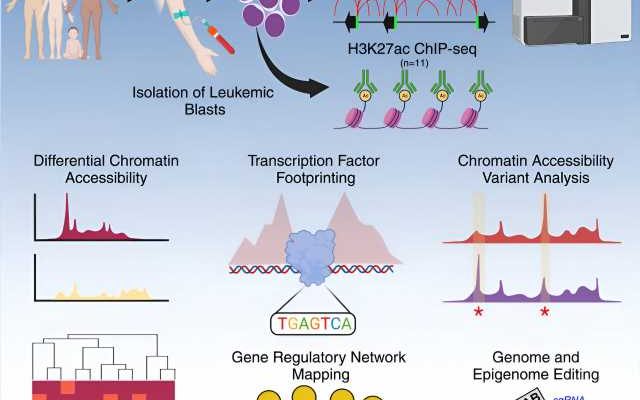
. DOI: 10.1016/j.xgen.2023.100442")
Scientists at St. Jude Children’s Research Hospital described the gene regulatory networks contributing to differences between subtypes of the most common childhood cancer, acute lymphoblastic leukemia (ALL). The work examined chromatin, the packaging that compacts DNA, in a cohort of patient samples six times greater than any previous efforts. The results serve as a valuable resource to better understand why patients’ outcomes differ and to ultimately improve treatments. The findings were published today in Cell Genomics.
“We wanted to understand how ALL subtypes differ from one another at the genomic level and answer the question, what makes a leukemic cell a leukemic cell and not a healthy cell?” said corresponding author Daniel Savic, Ph.D., St. Jude Department of Pharmacy and Pharmaceutical Sciences. “To do this, we mapped the activity of the non-coding molecular switches that control gene expression and contribute to the formation of gene regulatory networks in patient tumor cells and compared them to healthy cells.”
The majority of cancer research has focused on mutations that occur in the “coding” portion of the genome, the less than 2% of human DNA that contains genes. However, the “non-coding” genome portion, which accounts for the remaining 98%, is also important and contains instructions for controlling how and when genes are expressed. Previous research has shown that disruptions to proteins that influence the non-coding genome portion are prevalent in several types of cancer.
The St. Jude group, therefore, examined the non-coding genome to understand what made a cancer cell a cancer cell. To do this, the scientists measured the accessibility of chromatin, the packaging that compacts DNA in the cell nucleus. This determines which DNA pieces are available to be read in a particular cancer cell. These sets of DNA instructions and genes available to a cancer cell, known as gene regulatory networks, are key to understanding a cancer’s identity—and, potentially, its weaknesses.
“Certain subtypes of ALL don’t respond well to current treatments, and in many cases, we don’t understand why,” Savic added. “This resource could be used to identify the underlying gene regulatory differences playing a role in those heterogenous treatment responses. By taking into account these alterations to gene regulatory networks, in the future, we could theoretically modify or generate therapeutics to treat each subtype more effectively.”
Chromatin accessibility: The ‘window to the genome.’
Chromatin refers to DNA and the proteins that help to organize and package our genetic material efficiently. When chromatin is closed, it prevents gene expression. When chromatin is open, it makes DNA more accessible for gene expression and allows DNA to make three-dimensional structures regulating that expression. Gene expression is the process of using the information contained in a piece of DNA, a gene, to create a functional product such as a protein.
The St. Jude group evaluated how open or closed chromatin was—its accessibility—across the non-coding genome in 156 pediatric leukemia samples. Looking at accessible chromatin and the molecular switches or instructions residing there gave the investigators insight into the complex gene regulatory networks that determine cell type—including healthy versus cancerous cells.
“Chromatin accessibility is the window to the genome,” said first author Kelly Barnett, Ph.D., St. Jude Department of Pharmacy and Pharmaceutical Sciences. “Even though the genome is vast, 3 billion base pairs, we’ve narrowed down which areas are important to look at to understand pediatric ALL.”
Picture the human genome as a large house with windows. Some windows are completely closed, and others are fully open. Open windows allow people (transcription factors) inside to make changes; closed windows keep people out.
Chromatin accessibility analysis looks at which windows are open and allowing people inside—places of the genome that are open, active, and bound by transcription factors. The researchers found the thousands of windows that were open, documenting differences between healthy and cancer cells and, perhaps more importantly, between ALL subtypes.
Finding transcription factor footprints in ALL
The findings also included a comprehensive analysis of the footprints of transcription factors, or proteins involved in gene expression, by turning specific genes on or off. These footprints are the predicted places where the proteins regulating gene expression will likely bind open chromatin. Importantly, the St Jude team discovered that the pattern of transcription factor binding differed between subtypes, offering another way to understand the nuanced gene regulation differences in cancer subtypes.
The researchers found that looking at chromatin accessibility and transcription factor footprints combined from just 156 patient samples—identifying which windows were open—could be used to predict the leukemia subtype with 89% accuracy.
Conventional methods, such as RNA sequencing based on thousands of samples, have a 91% predictive accuracy, suggesting this new approach could improve subtype identification and, therefore, treatment approaches with higher sample sizes or when combined with conventional RNA sequencing approaches.
Superior samples achieve superior results
The maps the scientists generated are considered of the highest quality, generated from samples that are the gold standard in research. The group used biopsies of cancers taken from patients, which were transported to the lab as quickly as possible. These fresh samples are more representative of ALL than the cell lines used in earlier research.
“This big batch of data originating directly from patient samples is very valuable to the field,” Barnett said. “There’s a major gap for epigenomic data sets, such as chromatin accessibility maps, from direct patient samples. This study is our effort to fill that gap and provide a useful resource for the scientific community.”
More information:
Kelly R. Barnett et al, Epigenomic mapping reveals distinct B cell acute lymphoblastic leukemia chromatin architectures and regulators, Cell Genomics (2023). DOI: 10.1016/j.xgen.2023.100442
Journal information:
Cell Genomics
Source: Read Full Article