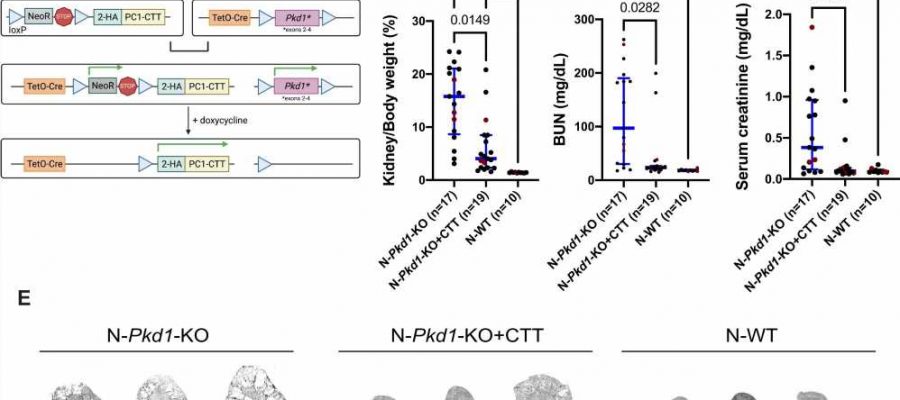
 suppresses cystic disease in an orthologous mouse model of ADPKD. A Design of the 2HA-PC1-CTT;Pkd1fl/fl;Pax8rtTA;TetO-Cre (Pkd1-KO + CTT) mouse model. We generated transgenic mice that carry a BAC-2HA-PC1-CTT transgene inserted in the Rosa26 locus and preceded by a neomycin resistance (NeoR) STOP cassette flanked by loxP sequences. These mice were crossed with the previously characterized Pkd1fl/fl;Pax8rtTA;TetO-Cre mouse model of ADPKD in which exons 2–4 of the Pkd1 gene are flanked by loxP sequences. Cre-mediated recombination of these second-generation 2HA-PC1-CTT;Pkd1fl/fl;Pax8rtTA;TetO-Cre mice via doxycycline induction promotes 2HA-PC1-CTT protein expression and loss of full-length PC1 protein expression in tubular epithelial cells. B–D Comparative analysis of N-Pkd1-KO, N-Pkd1-KO + CTT, and N-WT mice showing differences in KW/BW ratio (B), BUN (C), and serum creatinine (D). Cystic mouse cohorts are composed of 53–58% female and 42–47% male mice. Red dots represent the animals depicted in (E). E Representative H&E-stained kidney sections (4×) from 16-week N-Pkd1-KO, N-Pkd1-KO + CTT, and N-WT mice. Each group of sections is representative of the average KW/BW ratio of the entire cohort and is illustrative of the inherent variability associated with this mouse model. Scale bar: 2 mm. Non-parametric data are depicted with the median and interquartile range (blue bars). Multiple group comparisons were performed using Kruskal–Wallis test followed by Dunn’s multiple-comparisons test. H&E-stained kidney sections from all of the cystic mice included in this cohort are provided in Supplementary Fig. 1. Source data are provided as a Source Data file. Credit: Nature Communications (2023). DOI: 10.1038/s41467-023-37449-1")
Autosomal dominant polycystic kidney disease (ADPKD) is the most common potentially lethal genetic disease—about a half million people in the United States alone suffer from the condition. There is no cure, but new research could open the door to new gene therapies for treating most cases of the disease.
For several decades, researchers have known that mutations in the PKD1 gene, which encodes the polycystin-1 (PC1) protein, can cause the disease in about 80% of cases. However, the protein is too big to be modified through gene therapy strategies.
Now, a research team led by Laura Onuchic, MD, postdoctoral researcher in the Yale Department of Cellular & Molecular Physiology and Michael Caplan, MD, Ph.D., chair and C.N.H. Long Professor of Cellular & Molecular Physiology and professor of cell biology, has found that just a small piece of this protein might hold the key to preventing the disease. This finding could lead to opportunities to develop a new class of therapeutics. The team published its findings on March 30 in Nature Communications.
“Our research shows that a tiny fragment of the PC1 protein—just 200 amino acids from the very tail end of that protein—is enough to suppress the disease in a mouse model,” says Caplan, who was principal investigator of the study. “Our work will provide new insights into the underlying disease mechanisms for polycystic kidney disease and reveal new avenues for developing therapies.”
ADPKD is a genetic disorder that affects around one in 1,000 people. The affected kidneys develop cysts that grow in number and size. Each of our kidneys has around a million nephrons. In the disorder, over the course of decades, some of these nephrons develop into large, fluid-filled cysts that crowd out the normal tissue. Over time, this can compress and degrade the functional portion of the kidney, leading to loss of kidney function.
“By that time, a patient’s kidneys are very large—they can be the size of a football,” says Onuchic, who is first author of the study. In comparison, a normal kidney is around the size of a fist.
Around half of those with the disease will experience renal failure requiring dialysis or a kidney transplant. Furthermore, it can be passed from parent to offspring—if one parent is a carrier, half of their children are likely to be affected. “So, you have all these large families where multiple people carry the condition,” she says.
A little over a year ago, a team led by Stefan Somlo, MD, C.N.H. Long Professor of Medicine (Nephrology) and professor of genetics, found that if they removed the PC1 protein in mouse models, the kidneys became enlarged. After the protein was re-expressed, the kidneys returned to normal.
“They did a really beautiful experiment showing that in mouse models of polycystic kidney disease, where these animals get huge cysts in their kidneys, even when those cysts have already developed, turning the expression of the normal protein back on makes the cysts go away,” says Caplan.
“The problem with this as a therapeutic strategy is that this protein in 4300 amino acids long,” adds Onuchic. “It’s too big for gene delivery.” The solution, Onuchic and Caplan say, may be to bring gene therapy for ADPKD down to a manageable scale.
Researchers use gene therapy to try to take the sequence that encodes their gene of interest and get it expressed in their desired cells. This usually involves viral vectors. “Viruses can be the Trojan horses that deliver your gene of interest into the cell you need to get it into, but those viruses only have a certain amount of room in their trunk” says Caplan.
Because the PC1 protein is massive, this poses a problem for treating polycystic kidney disease. “PC1 is way too big to fit in the Volkswagen Beetle that is most gene therapy vectors, but now just this 200 amino acid piece can fit in the glove compartment.”
In their new study, the team used a mouse model that they had genetically modified to allow them to turn off the genes associated with polycystic kidney disease. In other words, they genetically induced the disease in these models by creating mutations in the genomes of the mice. As a result, the models developed cysts.
Then, the team turned on the expression of the 200 amino acid-long fragment of the protein. “Imagine flipping a light switch where one light goes off and one light goes on,” says Caplan. “We’re turning off the normal polycystic kidney disease gene and turning on the expression of just this tiny piece of the protein.”
The team found that this dramatically reduced the size of the cysts. “Even though we got rid of the full length PC1 protein, which would normally cause significant cystic disease, just turning on this tiny piece was enough to suppress the disease,” he says.
Furthermore, the team unveiled clues for the mechanism behind why this small piece is sufficient on its own. Through immunoprecipitation, they used an antibody to isolate the protein, and then used mass spectrometry to identify which proteins interact with it. They found that a mitochondrial protein called Nicotinamide Nucleotide Transhydrogenase (NNT) interacts with the PC1 fragment.
“From a basic biology point of view, this tells us something entirely new about what the polycystic kidney disease protein does and opens up a whole avenue to study its normal function,” says Caplan.
The team plans to continue pursuing the use of gene therapy, initially in mouse models, for just the 200 amino acid piece, with hopes that their work will one day benefit humans. “From a therapeutic perspective, it’s really exciting that we’ll hopefully be able to at least slow down disease progression,” says Onuchic.
More information:
Laura Onuchic et al, The C-terminal tail of polycystin-1 suppresses cystic disease in a mitochondrial enzyme-dependent fashion, Nature Communications (2023). DOI: 10.1038/s41467-023-37449-1
Journal information:
Nature Communications
Source: Read Full Article